Join us for an interactive 1-hour webinar on variant calling. We will cover:
TIPS ON DESIGNING YOUR EXPERIMENT
We will walk you through some of the more important questions to consider when planning your experiment. Should you go with WGS, WES, or a panel? What depth of coverage should you choose? And if you’re sequencing tumors, what is the purpose of a normal control or a panel of normals?
WHAT ARE SOME TOOL AND PARAMETER BEST PRACTICES?
We will provide a quick comparison of some variant callers and guide you on which you should choose depending on your needs. Moreover, among the dozens of parameters these tools have, we will talk about those you may want to change and how they could impact your analyses.
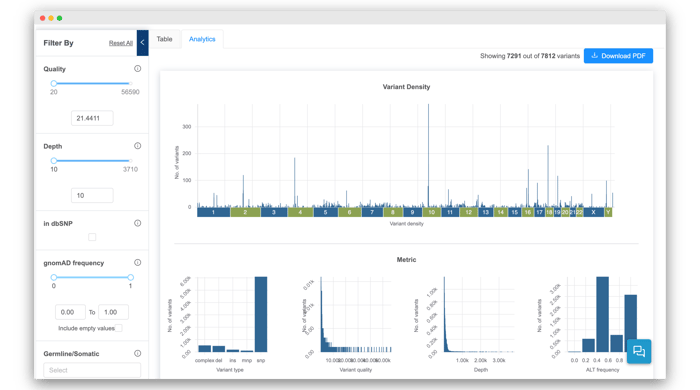
HOW TO TELL IF YOUR DATA IS POOR QUALITY, AND WHAT TO DO ABOUT IT
We will guide you through some key metrics and plots, including read quality metrics, mapping metrics, and coverage. You will learn about approaches you can take to mitigate poor quality data.
WAYS TO VALIDATE YOUR VARIANT CALLING RESULTS
How can you check if your variant calls are real? We will highlight key variant metrics to examine and show you how to use the genome browser.
.png?width=155&name=newlogo-svg%20(1).png "Basepair")